林道会议重要发言人之一的布赖恩·科比尔卡(Brian K. Kobilka)认为,了解G蛋白耦合受体的结构可帮助药物的设计开发,但这一切又是怎么进行的呢?
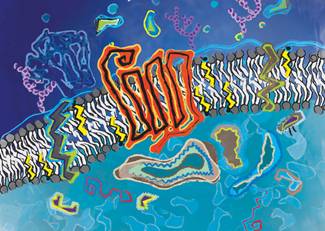
卡蒂亚·卡迪谢维斯基(Katya Kadyshevskaya )的斯克里普斯GPCR网发现的G蛋白耦合受体和它的七个跨膜区
医学上深有造诣的布赖恩·科比尔卡感觉,自己在今夏的林道会议上“就像一位刚加入化学俱乐部的新人”,但实际上,几乎每一位与会者对他获得诺奖的成就――G-蛋白耦合受体(GPCR)的研究发现都已经非常熟悉了。用美国北卡罗莱纳大学结构生物学家布赖恩·罗斯(Bryan Roth)的话说,这一研究成果“已经彻底改变了新的GPCR药物开发的途医学上深有造诣的布赖恩·科比尔卡感觉,自己在今夏的林道会议上“就像一位刚加入化学俱乐部的新人”,但实际上,几乎每一位与会者对他获得诺奖的成就――G-蛋白耦合受体(GPCR)的研究发现都已经非常熟悉了。用美国北卡罗莱纳大学结构生物学家布赖恩·罗斯(Bryan Roth)的话说,这一研究成果“已经彻底改变了新的GPCR药物开发的途径”,并可能为许多疾病的治疗提供新的方法。

2012诺贝尔化学奖得主科比尔卡
GPCRs可对体内几乎所有细胞发出的各种信号进行调节,包括荷尔蒙激素、神经递质、离子、气味分子,甚至还有光线。在被匹配的分子(或配体)激活后,GPCRs产生结构性变化,引发细胞的一连串连锁反应。以GPCR家族的不同成员为靶标开发的药物,可产生各种大的生理效应。例如,β受体阻滞剂能减缓心率、抗组胺药可以预防过敏反应、安定药可调节神经传递等。因此,GPCRs给药物设计带来了无限的可能性。事实上,正是GPCRs对于重症护理药物研发的重要性,吸引着科比尔卡走出临床门诊进入了实验室。
科比尔卡和罗伯特·莱夫科维茨(Robert J.Lefkowitz,2012年诺贝尔化学奖得主之一)的研究揭示了GPCR家族的常见结构及其变异成员。澳大利亚莫纳什大学的药理学家阿瑟·克里斯托泼罗斯(Arthur Christopoulos)认为,将生物学上的数据转换为药物发现上的进步,需要“生物学家和化学家的密切合作”。目前,很多研究人员都在致力于利用GPCRs的新的信息,以结束“钝锤”药物一统天下的局面。克里斯托泼罗斯也是其中之一。
这项研究已经超越了学科界限。研究人员希望设计的以GPCRs为靶标的新的药物,可提供更有效的治疗和更多的选择性,并尽可能地消除药物的副作用。正是基于这些新的知识,药物化学家开始设计绑定到某个特定受体上的新的配体,可比现有药物更有效地改变受体的活动。
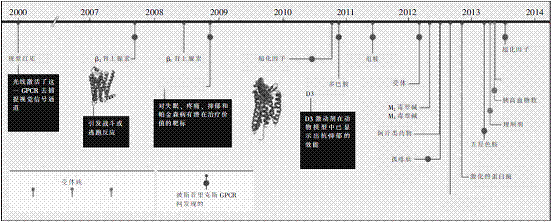
由于2008年建立的斯克里普斯GPCR网的努力,被破解的GPCR结构的数量正在迅速增加。斯克里普斯GPCR网是一个大型的网络协作项目,涉及十多个研究所和工业实验室,其目标是在2015年前至少确定15个 GPCRs的特点,目前已经破解了13个
GPCRs结构:药物开发的精确工具
药物研发是一个缓慢而昂贵的过程。进入临床试验第一阶段的药物,最终只有5%不到能够获得美国食品和药品管理局(FDA)的批准,每种药物的开发成本估计接近10亿美元。直到不久之前,人们对包括那些以GPCRs为靶标的大多数药物的构成结构和其生物学原理的理解还都十分有限。
GPCR药物开发最成功的例子是那些能通过如肾上腺素和复合胺等小分子激活的受体,如于45年前获得批准的肾上腺素受体显效药舒喘宁,至今仍然是治疗哮喘最有效的药物之一。但开发能与大的蛋白质或配体结合的有药物属性的小分子则是一个很大的挑战。由于GPCRs家族之间的相似性,很难鉴别选择的药物是否会产生意料之外的非目标行为。
如果研究人员能够反过来从受体结构入手,将有助于促进新药物的开发工作,即利用受体绑定的结构信息反馈到虚拟筛选程序中,通过识别每个受体绑定的特定区域,可以设计出具有高度选择性的配体,这也是靶向药物开发的第一步。“这些方法将导致候选药物高度优化并提高药物开发的效率。”英国靶点药物开发公司Heptares Therapeutics首席科学官菲奥纳·马歇尔(Fiona Marshall)说道。
马歇尔认为,GPCRs将逐渐成为一些罕见疾病治疗药物的开发目标(对于这类疾病目前几乎无任何有针对性的药物)。所谓的“孤儿受体”(即GPCRs的自然配体目前还属于未知)也是具有吸引力的药物靶标。Heptares正在对最近摆脱了“孤儿受体”之名的受体进行研究,确认了GPR39的促效药(GPR39参与胰岛细胞功能,一直是糖尿病治疗的目标之一)。马歇尔解释说:“这种受体特别的意义在于,它有可能阻止糖尿病的发展。”此外,Heptares公司正在与其他制药公司合作,使用稳定的纯GPCR制剂产生的抗体激活或抑制受体活动,这类治疗性抗体药物具有许多优势。“一些具有特异性的抗体作用持久,可与其他形式的药物配合用于癌症治疗。”马歇尔说道。一些公司开发的许多类似的抗体已经用于临床,例如,东京一家生物科技公司开发的药物mogamulizumab(商品名为Poteligeo),在日本已被批准用于治疗成人T细胞白血病。
GPCRs研究:改变世界医学的未来
结构研究已揭示了GPCRs在其他层次的复杂性。除了内源性配体绑定位点之外,GPCRs还有可以绑定其他分子的变构位点(变构位点的结构更为多样化),给GPCRs的微调提供了更多机会。变构位点的效应难以量化,尤其是对于许多GPCR的靶标来说,达到最佳治疗效果的信号水平还属于未知。但这是一个激动人心的领域,“如果我们能够了解受体信号与临床效果之间的联系,我们将能有更好的办法让药物拥有我们希望达到的属性。”澳大利亚莫纳什大学药理学家帕特里克·塞克斯顿(Patrick Sexton)解释道。
目前已有两种变构配体批准作为治疗药物,美国加州Amgen公司开发的cinacalcet(商品名为Sensipar),作用是激活钙敏感受体,用于治疗甲状旁腺功能亢进;英国ViiV Healthcare公司开发的maraviroc(商品名为Selzentry或Celsentri),用于阻断艾滋病病毒借以进入宿主细胞的趋化因子受体,以及许多小公司正在研究GPCRs的变构调节作用,用以治疗神经退行性疾病和精神疾病。瑞士Addex制药公司正在二期试验中的化合药物,通过逆转左旋多巴诱发的运动障碍治疗帕金森氏疾病患者,对于治疗极为罕见的肌张力障碍也有很大的价值。“与正位结合位点相比,这些化合物在受体亚型上拥有更大的选择性。”Addex制药首席执行官蒂姆·戴尔(Tim Dyer)说道。
在实验室里可通过将化合物同时作用于正位结合位点和变构结合位点进行药物筛选,虽然这样的双跨膜(bitopic)配体在实验室里已经确定,但都还没有进入临床试验。克里斯托泼罗斯指出,双跨膜配体的研究仍处于早期阶段,最重要的挑战之一是如何找到最好的办法,将与正位位点和变构位点结合的配体连接起来,这样结合的分子有可能“因太大而不能被适当优化并作为潜在的药物靶标而加以开发。”
马歇尔指出,基于GPCR结构的药物发现和开发“还处于早期阶段”,很多靶标难以结晶,更重要的是,这些GPCR结构在不停的变化之中。“在药物设计计算方法中,应将GPCRs的动态特性考虑进去,”克里斯托泼罗斯指出,“否则我们也将遇到其他药物开发同行在过去曾遇到过的药物开发成功率低的问题。”
但塞克斯顿乐观地预测,未来五年里,受体成功结晶的数量和用来描述反应的定量工具的数量都会增加。他补充说,“这些都将逐步对候选药物的选择产生影响。”随着生物学和化学领域学科交叉的继续,科比尔卡在化学俱乐部里很快将不再是一位新人了。
资料来源 Nature
责任编辑 则 鸣
● 相关链接 ●
“尤里卡”时刻:预示伟大发现的小发现
1968年,罗伯特·莱夫科维茨用放射性碘分子对一些荷尔蒙激素做了标记后发现,这些激素可以绑定细胞外的受体,然后触发细胞内的反应,首次证明了生物学活性受体的存在。
布莱恩·科比尔卡于上世纪80年代加入莱夫科维茨的研究团队,他们的研究目标是要分离出肾上腺激素受体的基因编码,肾上腺激素所起的一个关键作用是调节心血管功能。1986年,他们成功克隆了β2-肾上腺激素受体,发现它与视网膜紫质受体十分相似,其相似性表明,β-肾上腺激素受体是一个更大受体家族的一部分,它们在结构上相似,但拥有完全不同的功能。这些受体很快被命名为G蛋白耦合受体(GPCRs)。
在此之后,800多个GPCRs基因编码相继被确认,其中23个的结构已被破解。大多数这些受体的结构为A族(与视网膜紫质相似),包括肾上腺素、毒蕈碱和阿片受体,是治疗心脏病、呼吸停止和镇痛药物的既定目标。但GPCRs难以分离和结晶,它们不仅难以溶解,而且其结构也极不稳定,随着与不同配体和细胞内信号传输蛋白的交互作用而发生变化。直到2011年,科比尔卡设法让人体β2-肾上腺受体绑定于某个配体或G蛋白时被激活的那一刻(结晶),并同时给细胞发送信号。
最近已有来自不同家族受体结构的相关报道,包括SMO受体(smoothened receptor)和B族的两个受体――促肾上腺皮质激素释放因子1受体(CRF1R)和胰高血糖素受体。已得到验证的这些受体的临床目标疾病包括癌症和Ⅱ型糖尿病等,但由于它们与配体结合的表面较大,传统方法难以生成能够对它们进行攻击的小分子药物。
(“尤里卡”为希腊语,意思是“我知道了!我找到了!我想到了!”――译注)